Rachael Mansbach
Assistant Professor
re.mansbach@concordia.ca
Proteins are the building blocks of living things, miniature motors that make all of your cells function. Proteins embedded in cell membranes filter out toxic materials or uptake necessary nutrients. Meanwhile, malfunctioning proteins are responsible for a slew of disorders, including Alzheimer’s, type II diabetes and Parkinson’s.
Our group works on understanding and designing small molecules and peptides for therapeutic applications such as finding new analgesics for treatment of chronic pain disorders or correcting dysregulation of proteins that lead to Alzheimer’s.
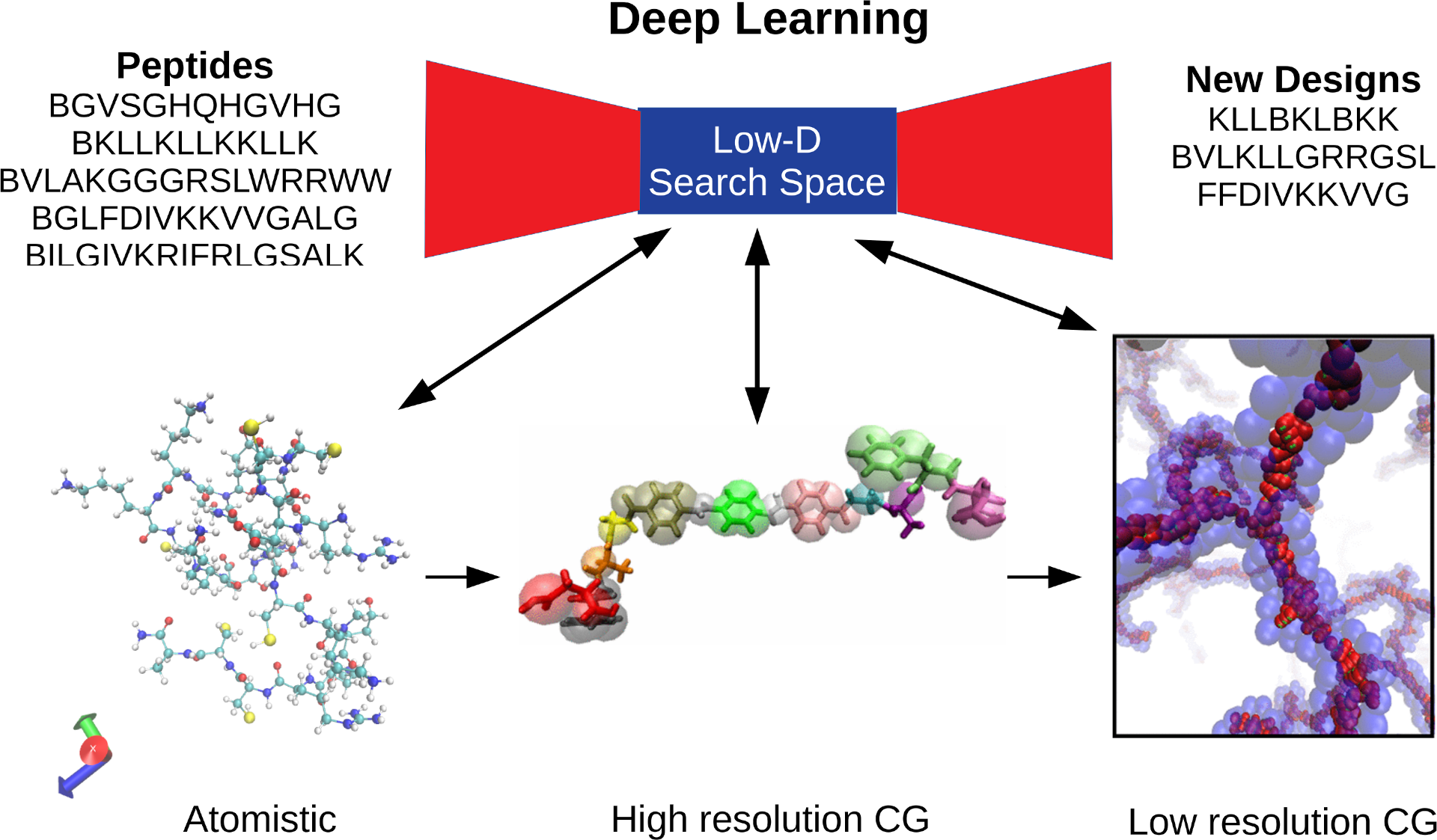 Schematic of deep learning procedure for therapeutic peptide design. Deep learning is used to produce a low-dimensional search space in which peptide capabilities are evaluated at different levels of resolution through molecular simulation and coarse-grained molecular simulation. High resolution CG and low resolution CG images from [1] and [2].
Schematic of deep learning procedure for therapeutic peptide design. Deep learning is used to produce a low-dimensional search space in which peptide capabilities are evaluated at different levels of resolution through molecular simulation and coarse-grained molecular simulation. High resolution CG and low resolution CG images from [1] and [2].
My primary scientific interests lie in designing small molecules and peptides (short proteins) for therapeutic applications: killing bacteria and drugging human cells in beneficial ways — for example to find new analgesics for treatment of chronic pain disorders, or to correct dysregulation of pathways involving the intrinsically disordered proteins that lead to Alzheimer’s. In order to do this, I employ theoretical and computational biophysics tools. I am particularly interested in the burgeoning field of deep learning as applied to molecular dynamics and drug design.
Over the past decade, generative deep learning (training an AI to create by pattern-matching to a large amount of data) has demonstrated a fascinating ability to create images and text, and there has been an explosion in the past three years over its application to drug design and molecular dynamics, but it still suffers from lack of interpretability and scalability. As a physicist, I am particularly interested in the interpretability issue.
With dwindling drug leads and increasing fatalities from multidrug-resistant bacteria, we are teetering on the brink of the post-antibiotic era. New design techniques and approaches for both understanding of antibiotic resistance and design of novel therapeutics are urgently needed. Recently, there has been promising experimental work in the field of antibiotic hybrids, in which sets of different pharmacophores are linked in novel ways, which may lead to molecules with enhanced efficacy or even entirely novel properties.
I am looking for a student interested in exploring the use of a fragment-based approach for novel antibiotic hybrid design through generative deep learning, in which a library of fragments relevant to antibiotic applications will be used as a basis for a generative model that produces novel antibiotic hybrids through linking, growth and merging, with a particular emphasis on interpretability as well as candidate generation.
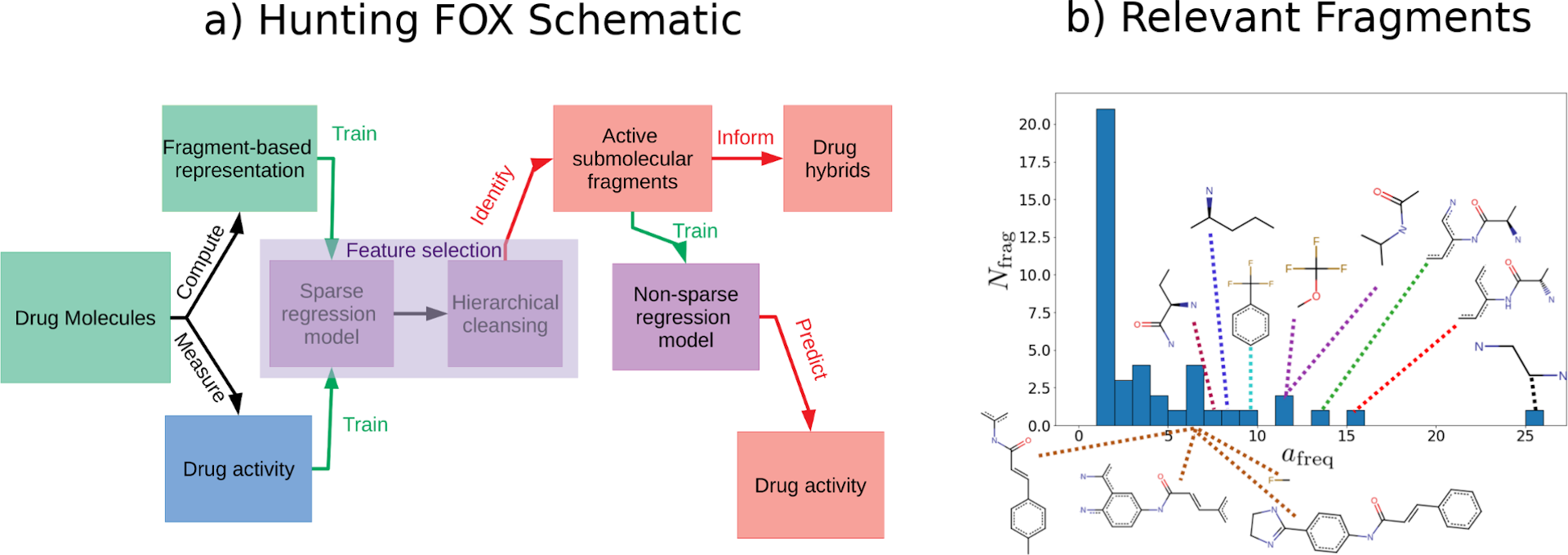 (a) Schematic of an early proof-of-concept method [1] ("Hunting FOX" for "Hunting Fragments Of X") for identifying fragments with relevant properties and (b) histogram of fragments identified by running the procedure in (a) 28 times on random splits of train/test data. The more often a fragment is identified, the more important it is. Figure modified from [1].
(a) Schematic of an early proof-of-concept method [1] ("Hunting FOX" for "Hunting Fragments Of X") for identifying fragments with relevant properties and (b) histogram of fragments identified by running the procedure in (a) 28 times on random splits of train/test data. The more often a fragment is identified, the more important it is. Figure modified from [1].
Although design of novel antibiotics is one approach to combat the growing problem of multidrug resistance, it is unlikely to be sufficient on its own and a multipronged approach is warranted. Another promising line of research for novel drugs with antimicrobial action is the design of peptides with antimicrobial properties that preferentially disrupt the membranes of bacterial cells while displaying a lower affinity for mammalian cells. This is still an active area of research, with only a few candidate peptides having reached clinical trials, due to the continued existence of unwanted side effects such as serum binding or the requirement of potentially-toxic concentrations to be effective.
I’m looking for a student interested in designing a deep learning model informed by multiscaled molecular dynamics, wherein generative learning is used to iteratively create potential AMP candidates that are assessed on multiple scales using molecular dynamics, from high-resolution atomistic molecular dynamics to assess detailed properties on short time scales, to low resolution “spherical peptide in a vacuum” models to assess penetration of candidates into the cell membrane.
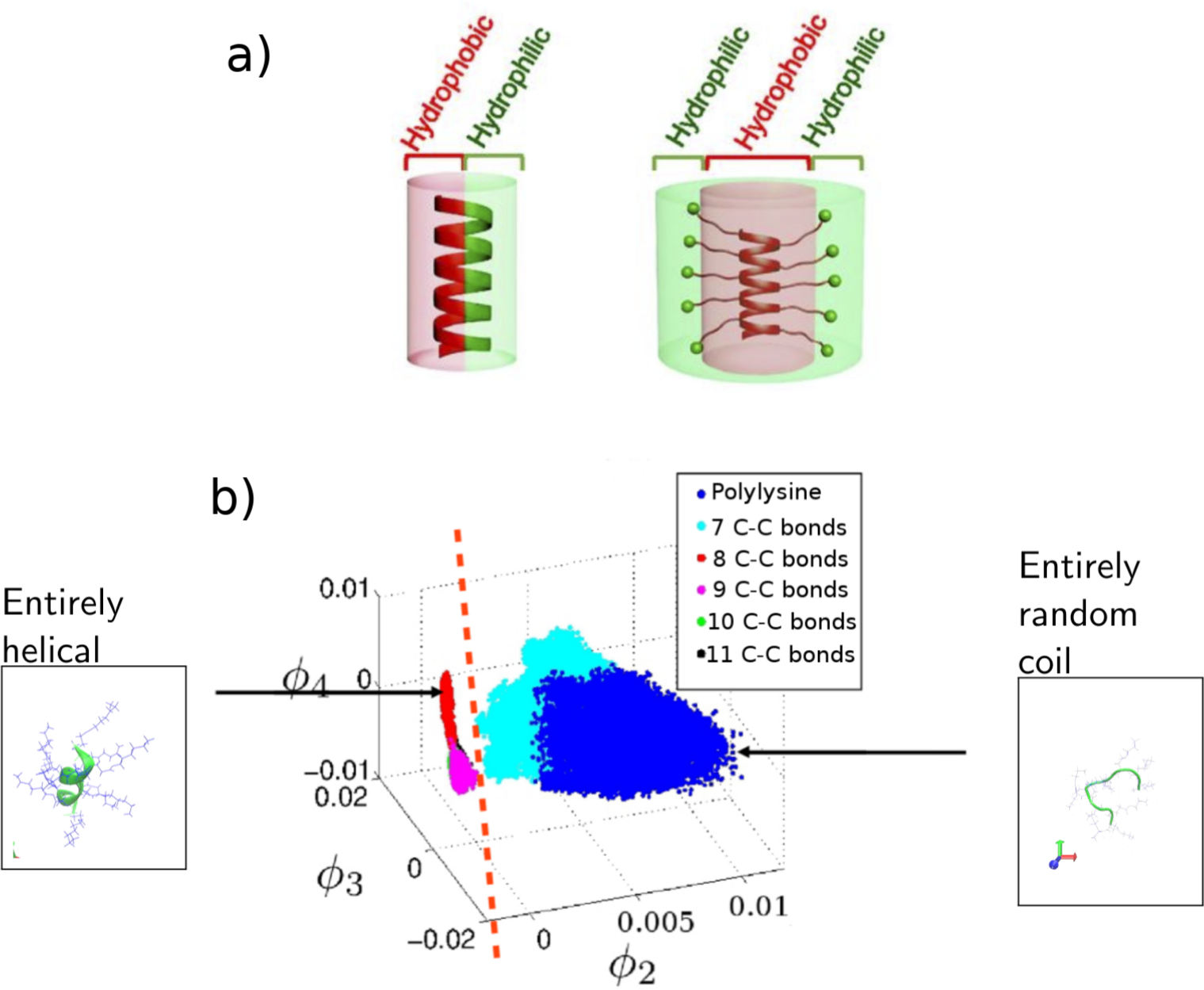 (a) Example of typical natural AMP -- hydrophobic on one side, hydrophilic on the other, versus an engineered AMP with radial amphiphilicity to improve performance. (b) Example of the use of unsupervised machine learning to map out the landscape of the engineered AMP to understand how to use the length of the side chains to change the helicity of the backbone. Figure modified from [1] and [2].
(a) Example of typical natural AMP -- hydrophobic on one side, hydrophilic on the other, versus an engineered AMP with radial amphiphilicity to improve performance. (b) Example of the use of unsupervised machine learning to map out the landscape of the engineered AMP to understand how to use the length of the side chains to change the helicity of the backbone. Figure modified from [1] and [2].
Owing to their high specificity and binding affinity for various receptors involved in different biological pathways, toxins have for a long time been considered a rich natural source of therapeutic leads. A large and intriguing class of toxins is those that are short, cysteine-rich peptides whose structures are largely controlled by the multiple disulfide bonds that form between the cysteines. Historically, one difficulty in assessing the structure and hence the proposed mechanisms of such toxins has been that it can prove difficult to controllably fold them to their native stable states in vitro. Indeed, there is evidence that certain toxins exist even in vivo in metastable states that are nonetheless potent and in certain cases the “non-native” fold demonstrates higher selectivity and binding affinity which leads to the question of whether we can control these metastable states through sequence, environment or kinetic engineering.
I am looking for a student interested in either polymer theory or molecular dynamics, to explore ways to bring together constrained polymer theories with bond-breaking force field models to map out the free energy landscapes of disulfide-rich toxins.
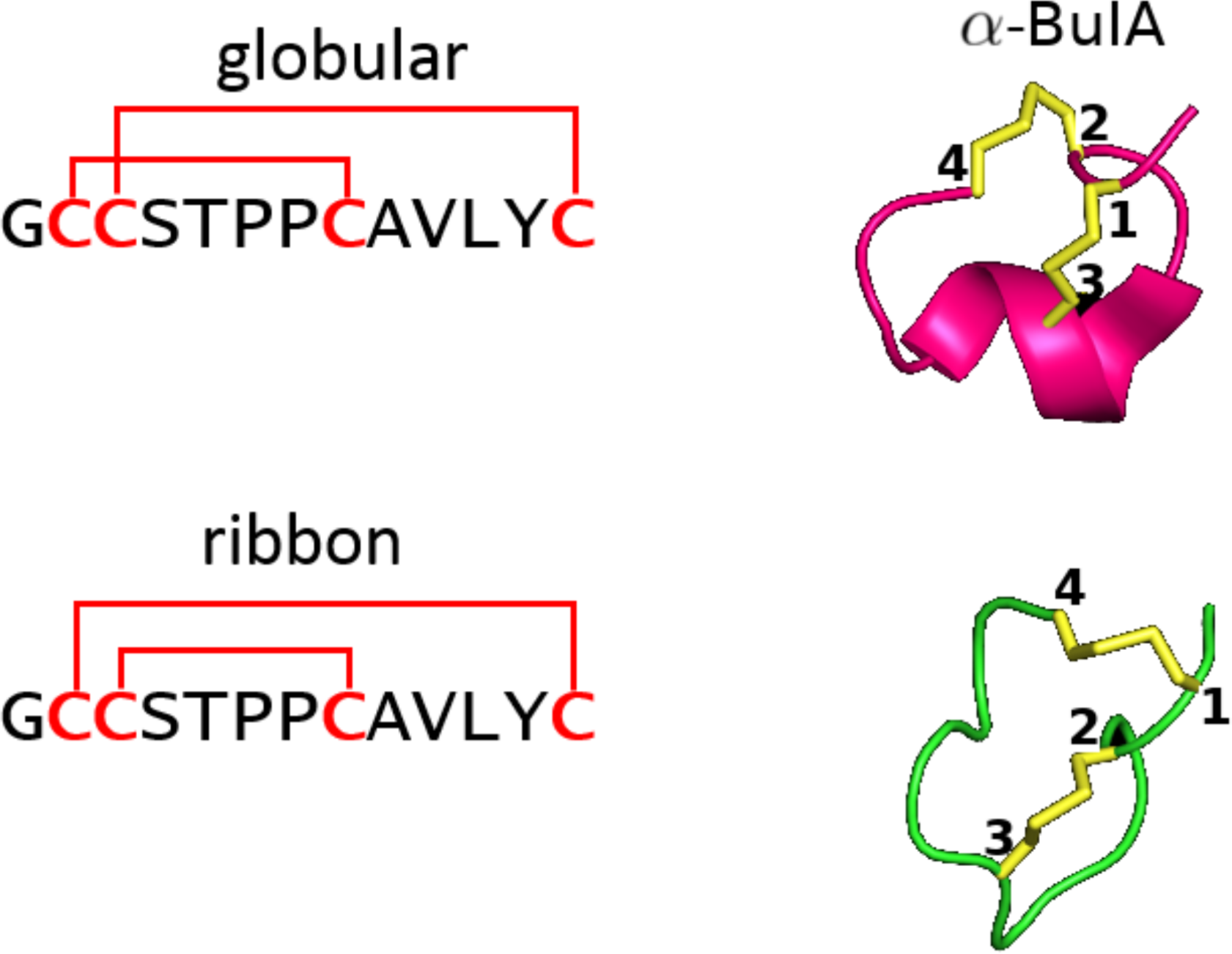 Example of two very different configurations ("globular" and "ribbon") taken on by alpha conotoxin AuIB when the cysteine connectivity is changed. Figure modified from [1].
Example of two very different configurations ("globular" and "ribbon") taken on by alpha conotoxin AuIB when the cysteine connectivity is changed. Figure modified from [1].

Assistant Professor
re.mansbach@concordia.ca

Los Alamos National Lab

University of Oklahoma
(Center for Antibiotic Discovery and Resistance)

Los Alamos National Lab

Los Alamos National Lab

Los Alamos National Lab

Los Alamos National Lab and Pebble Labs
MSc student
Please contact Rachael Mansbach if you have any questions or need their assistance.
Rachael Mansbach
Department of Physics
7141 Sherbrooke St. W.
Montreal, Quebec
H4B 1R6, Canada
© Concordia University